

SOP for Reporting and Handling of Deviation to ensure timely and effective reporting and handling of deviations in pharmaceutical processes to maintain product quality and compliance with regulations
Purpose
To describe procedure for reporting and handling of deviation(s)
Scope SOP for Reporting and Handling of Deviation
The scope of this document pertains to all the departments responsible for carrying activities related to the manufacturing of the products at Pharmaceuticals.
Responsibilities
- It is the responsibility of QA to prepare, manage and properly implement this SOP
- It is the responsibility of all the departments to intimate QA prior to do or immediately after occurrence of any deviation from the written approved procedure
- It is the responsibility of QA to trigger relevant department to intimate upon finding any deviation from written approved procedure
- It is the responsibility of personnel of concerned department to notify immediately their supervisor when any deviation occurs or notices
- It is the responsibility of QA to verify the deviation and action to be taken
- QA ensures that this SOP is followed in its entirety SOP for Reporting and Handling of Deviation
- It is the responsibility of QA Documentation Officer to prepare and update this SOP
- It is the responsibility of Assistant QA Manager or Designee to review this SOP
- It is the responsibility of QA Head or Designee to approve this SOP
- It is the responsibility of Director Technical to authorize this SOP
Definitions & Abbreviations
- Deviation: Any action of departing from an already approved written procedure
- Critical attribute: The critical attribute is one that defines and contributes to safety, identity, purity, strength or quality of a product, process or system
- SOP: Standard Operating Procedure
- QM: Quality Management
- BMR: Batch Manufacturing Record
- BPR: Batch Packaging Record
- cGMP: Current Good Manufacturing Practices
- HOD(s): Head of Department(s)
Departmental Codes
- QA: Quality Assurance
- QC: Quality Control
- PR: Production
- EG: Engineering
- WH: Warehouse
Procedures SOP for Reporting and Handling of Deviation
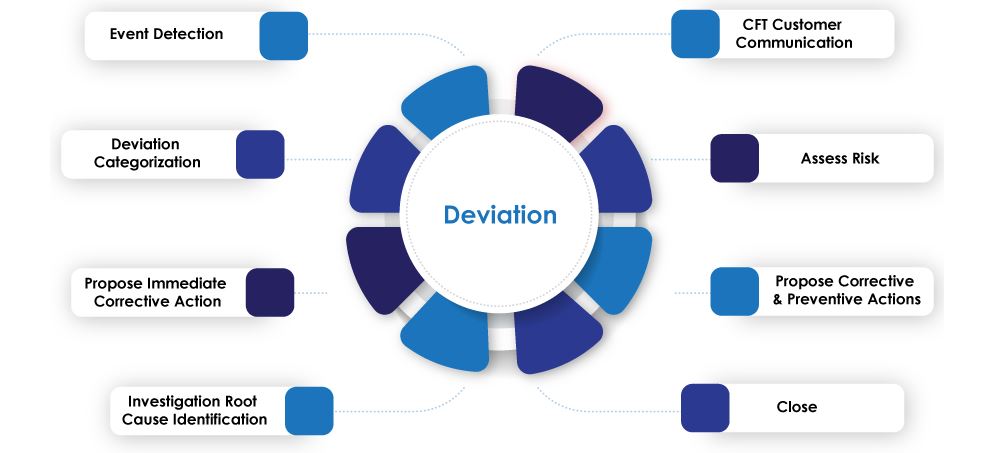
Classification of Deviation
Deviation is classified into two main categories
- Planned Deviation: Any deliberate or intentional non-conformance or deviation from established and approved procedures, planned prior to execution of an activity
• No critical deviation which has potential to alter the quality of product is to be planned
• Planned deviation is carried out only for but not limited to - Quality improvement
- Yield improvement
- Safety reasons
- Break downs
- Better GMPs
- Market requirement
- Insufficient inputs
Example: Use of Fitz mill instead of Granulator in Production
- Unplanned Deviation: An accident or unanticipated non-conformance or deviation from approved written procedure observed or noticed during or after execution of activity
• An unplanned deviation can be critical, major, or minor in nature
• The unplanned deviation related to any procedures, processed, system and utilities
• These deviation may occurs for any reason, such as following (not all inclusive) - Equipment failure/ breakdown, Malfunctioning
- Power supply failure (This is an exception in Pakistan due to power shortage, happened several times on daily basis. Power generators are on stand by and kick into effect)
- Breakdown in support service / utilities
- Human error
- Documentation Error
- Laboratory Failure
- Yield deviation
Example: Failure in procedure, utility, material, equipment, or any system is occurred
Planned (Major / Minor) and Unplanned (Critical / Major / Minor) deviations are sub classified on the basis of their impact on product quality
A. Critical Deviation: The deviation that has a direct impact on critical attribute of product, process or system as well as impact on GMP
Example: Manufacturing instructions are not followed. Wrong batch details are printed. SOP or methods of testing not followed during analysis
B. Major deviation: The deviation could or may possibly has an impact on critical attributes of the product as well as impact on GMP
Example: Raw material is received in damaged containers
C. Minor Deviation: The deviation is unlikely to has an impact on critical attribute of the product and nor unlikely to has impact on GMP
Example: Line clearance is not taken from QA, Physician sample wrongly printed with price etc.
Planned and Unplanned deviations are further classified as;
A. System Deviation: Any under or overshoots of critical system (i.e. HVAC, Water, Pressure, Electricity etc.)
B. Process Deviation: Any planned or unplanned deviation related to a process or operation from already established criteria (i.e. Drying in FBD instead of Tray Dryer)
Note 1: The Deviations related to Facility, Equipment, Batch Size etc. will be mentioned in category of others
Note 2: No critical planned deviation is allowed which has potential to alter the quality, purity or safety of the product
Note 3: Critical and major deviations can be taken and implemented ONLY after proper evaluation, risk investigation and pre-approval from Quality Council members and Director Technical / Operations
Initiation of Deviation Process
Deviation (Planned / Unplanned) can be initiated as a result of following but not limited to
- Any relevant department / regulatory authority finds occurrence of deviation
- QA finds out any deviation from approved procedure
- Service Requests, Internal Complaints, External Complaints
- Self-inspections, Product Quality Reviews
- Supplier / Service provider communications
- Process monitoring / Environmental monitoring
- SOP for Reporting and Handling of Deviation
HOD or System Owner accesses the proposal of planned / unplanned deviation, justification given for its potential impact on the product quality, compliance to regulatory requirements, GMP and advise on root cause and Corrective & Preventive action (CAPA)
After review by HOD and completion of Section A, the Deviation Form SOP for Reporting and Handling of Deviation (QA/FRM/0000/00) is forwarded to QA for review and approval
Deviation Investigation SOP for Reporting and Handling of Deviation
- QA issues deviation number and records into Deviation Log Book
- QA head reviews the Deviation Form with respect to impact on product quality, feasibility of planned / unplanned proposal, rational / justification and compliance to GMP, the root cause and CAPA suggestions along with members of Quality Council and Director Technical / Operations where necessary
- QA approves / rejects the planned / unplanned deviation with appropriate recommendations
- QA must gather all facts concerning deviation and provide an assessment of deviation on product quality
- After approval from QA Head, QA allows the relevant department for further proceeding
- The concerned department is to implement the planned / unplanned deviation
- The observations and data generated must be documented
- Where necessary, the affected batch must be identified, removed from production and placed under Quarantine with Hold label by QA till investigation is completed and decision is made by QA
- Perform risk assessment where necessary as per SOP for Quality Risk Management
- On the basis of investigation, status of batch must be determined and recorded in deviation form
- CAPA should be taken to prevent reoccurrence of deviation in future
Note 1: Planned deviation must acquire prior approval from Director Technical / Operations
Note 2: In case of Critical (Unplanned) Deviation, batch must be on HOLD until final decision is made
Deviation Tracking Numbering System
QA assigns a unique number to each planned / unplanned deviation as given below;
DEV / DD / YY / 0000
where;
DEV: (Planned / Unplanned) Deviation
DD: Departmental Code
YY: Year in which deviation is initiated and recorded
0000: Sequential Number of deviation in a calendar year
Example: DEV / PRD / 24 / 0001 is the First deviation of Production for the year 2024
QA maintains a logbook for the approved planned / unplanned deviation (Annexure A)
Closure of Deviation
- The closure of planned / unplanned deviation is the joint responsibility of Heads of relevant department and QA
- QA reviews implementation and document data obtained from respective department to ensure that the recommendations are complying with quality profile of batch(s) impacted by planned / unplanned deviation
- QA is to close the deviation by reviewing and assessing the impact of planned / unplanned deviation on the quality of the product as well as its impact on GMP
- The time allowed for handling and closing planned and unplanned deviation should not be more than a month
- In case the deviation is not closed within a month, it must be properly investigated and justified by HOD of relevant department
- Deviation must be closed before the release of specific batch
- SOP for Reporting and Handling of Deviation
Note 1: The effectiveness and follow up of the deviation / action taken and trending to see if the same events are repeatedly occurring must be monitored and discussed in Quality Council Meeting
Note 2: If the planned / unplanned deviation is related to process / procedures that lead to improvement in the product quality, then the planned / unplanned deviation can be made permanent by following the change control procedures as defined in SOP for Change Control (QA/SOP/QM/0000)
Attached Documents (Forms / Annexure)
- Flow Diagram for Handling of Deviation (Annexure A)
- Format for Deviation Log Book (Annexure B)
- Deviation Form (QA/FRM/0000/00)
Reference & Linked Documents
- WHO Guidelines on Deviation Handling
- ICH Guidelines, Quality Risk Management Q9
- MHRA Guidance on GMP Chapter – 1, Quality Management
- SOP for Management of Documents
- SOP for Quality Council
- SOP for Change Control
- SOP for Quality Risk Management
- SOP for Corrective Action & Preventive Action
Pretty nice post. I just stumbled upon your weblog and wished to
say that I have really enjoyed surfing around your blog posts.
In any case I’ll be subscribing to your feed and I hope you write again very soon!
I loved as much as youll receive carried out right here The sketch is attractive your authored material stylish nonetheless you command get bought an nervousness over that you wish be delivering the following unwell unquestionably come more formerly again as exactly the same nearly a lot often inside case you shield this hike
thank you so much
Logar açmaÜmraniye Ümraniye Lavabo Tıkanıklığı Açma İstanbul-Ümraniye sıhhi tesisatçısı olarak İstanbul Ümraniye bölgesinde 7 gün 24 saat su tesisatçı hizmeti sunmaktayız.Lavabo açma hizmetimiz son teknolojik cihazlarla kırmadan dökmeden siz değerli müşterilerimize 7 gün 24 saat hizmet vermenin onur ve mutluluğunu yaşıyoruz.Artık Lavabo Açma çok kolay sizleri büyük sıkıntıdan anında kurtarıyoruz. https://hksinstruments.com/2024/03/31/umraniye-mutfak-tikanikligi-acma/
thank you so much
I truly relished the effort you’ve invested here. The design is tasteful, your authored material fashionable, however, you seem to have acquired some unease about what you intend to present henceforth. Undoubtedly, I’ll revisit more regularly, similar to I have nearly all the time, in the event you sustain this rise.
Ümraniye televizyon montajı Vaviyen anahtar terimi, dilimize İngilizce “Vavien key” ifadesinden geçmiş, evsel kullanım için sıcak ve soğuk su musluğunu açıp kapatan, sıcak suyun aniden gelmesini önleyen ve bu sayede suyun enerji tasarruflu kullanılmasını sağlayan bir musluk tertibatını ifade eder. https://celebisland.com/read-blog/1468
thank you
What a fantastic resource! The articles are meticulously crafted, offering a perfect balance of depth and accessibility. I always walk away having gained new understanding. My sincere appreciation to the team behind this outstanding website.
The breadth of knowledge compiled on this website is astounding. Every article is a well-crafted masterpiece brimming with insights. I’m grateful to have discovered such a rich educational resource. You’ve gained a lifelong fan!
thank you so much