
Internal Audit in Pharmaceutical Industry SOP
Internal Audit in Pharmaceutical Industry SOP is based upon standard FDA guidelines which are applied on every pharmaceuticals manufacturing industry.
PURPOSE
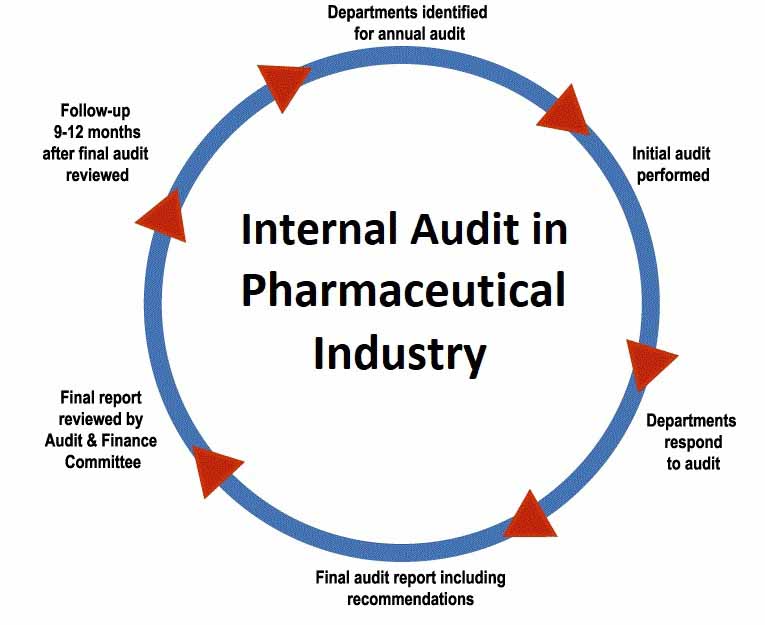
1.1. To describe procedure for planning, execution, reporting and follow up of cGMP Internal Audit in Pharmaceutical Industry.
SCOPE
2.1. This c.GMP audit procedure is applicable to all departments involved in Manufacturing, testing and storage of products.
RESPONSIBILITIES
3.1. It is the responsibility of Quality Assurance department to schedule cGMP internal audits biannually.
3.2. It is the responsibility of internal auditors to conduct audits as planned, prepare report and do follow up inspection to evaluate corrective and preventive actions.
3.3. It is the responsibility of the audited department to provide full access to its processes, procedures and systems for internal audit.
3.4. It is responsibility of the audited department to respond the audit within 15 days of the receipt of internal audit report.
3.5. It is the responsibility of QA manager to review and update internal audit procedure as necessary and make sure its compliance.
DEFINITIONS & ABBREVIATIONS
4.1. Current Good Manufacturing Practices (cGMP): Set of guidelines to ensure safety, quality, efficacy, identity and purity of manufactured products.
4.2. Internal Audit: A system to monitor the implementation and respect of good manufacturing practice and to propose any necessary corrective measures. Records of such inspection and any subsequent corrective action shall be maintained.
4.3. NCR: Non-conformance report.
MATERIALS & EQUIPMENTS
Not use
PROCEDURE
Auditor Selection
6.1.1.1. Internal auditors are selected by QA Manager from different departments
on the basis of their training and experience in cGMP implementation.
6.1.1.2. Attached form, FQA – 013, contains details of internal auditor team.
Planning And Scheduling Internal Audit
6.2.1.1. Audit all the departments involved in manufacturing, testing and storage
of products biannually
6.2.1.2. QA prepares schedule for audits and communicates it to all the
concerned departments, before time, through email and takes receiving on a hard copy.
6.2.1.3. In case of any change in schedule, the new schedule is communicated to
the auditor(s) and the audit department.
Audit Execution
6.3.1.1. Conduct audit during routine working hours.
6.3.1.2. Auditors must have a general audit plan including the following:
A) Previous audit report, if the department has been audited before.
B) Audit checklist.
6.3.1.3. Verify status of the previous audit report including recommendations.
6.3.1.4. Work in cooperation with the department personnel. It is also important to
get as much information as possible about the current procedures by;
a. Asking open-ended questions
b. Listening and understanding
c. Restating responses
d. Allowing time for responses
e. Showing flexibility and a constructive attitude
f. Choosing proper vocabulary
6.3.1.5. Verify that all documents, procedures and instructions are followed and implemented properly as described in authorized documents i.e. SOPs.
Non-Conformance Report
6.4.1. The findings of the internal audit are documented on the Non-Conformance Report, FQA – 011.
6.4.2. The non-conformances are classified into the following classes:
A) Critical: cGMP non-conformances that directly impact product quality, safety, purity and identity are categorized as critical, e.g. incorrect weighing of raw material.
B) Major: cGMP non-conformances that may impact product quality, safety, purity and identity, e.g. Use of un-calibrated equipment.
C) Minor: cGMP non-conformances that does not directly impact product quality, safety, purity and identity, e.g. Documentation issues: i.e. over writings or cross outs and corrections without initial and date.
6.4.3. The Non-Conformance is categorized as Critical, Major, Minor or Observation.
6.4.4. The auditee identifies and describes the root cause of the Non-Conformance and gives the target date to complete the corrective actions.
6.4.5. QA issues a sequential number to each Non-Conformance Report.
Audit Reporting
6.5.1. After completion of the audit, an audit report is prepared by the auditors, highlighting the state of cGMP compliance of the auditee department.
6.5.2. The audit report is given a sequential number as described below:
DD – MM/YY
DD = Department Code
MM = Month
YY = Year
Example = QC — 07/14
6.5.3. Send a copy of cGMP audit report to the auditee department while original is kept at QA department for record and follow up.
Audit Follow Up
6.6.1. Conduct follow up audit for each non-conformance to assess the action taken within target dates.
6.6.2. Write up a new non-conformance report with the same non-conformance number, if corrective actions have not been taken against non-conformance as per agreed target date.
6.6.3. Keep all GMP non-conformance reports and related documents in the QA file for five years.
Result
6.7.1.1. The result of Internal Audit Report is presented at the management review meeting.
6.7.1.2. Internal audit findings, reports, follow up actions are confidential information and is intended for internal use only and not subjected to external use.
6.8. Updating Audit Checklist To cGMP
6.8.1.1. QA updates audit checklist whenever required due to any changes in the national and international regulations.
REFERENCES
7.1. Good Manufacturing Practices (cGMP) for Manufacturers.
7.2. Schedule B-II under the drugs (licensing, registering and advertising) rules 1976, framed under the drugs act, 1976.
REQUIRED DOCUMENTS
NA
ATTACHED DOCUMENTS
9.1. cGMP Audit Schedule, FQA-010
9.2. Non-Conformance Report, FQA-011
9.3. Internal Audit Summary Report. FQA-012
9.4. cGMP Auditor Team, FQA-013
LIST OF RECIPIENTS
QA/QC Manager
Production Manager
Warehouse In-charge
Engineering Department