

A Standard Operating Procedure (SOP) for Out of Specifications (OOS) Handling Procedure results in the pharmaceutical industry is crucial for managing deviations from established specifications during the testing or manufacturing of pharmaceutical products. Below is a general outline for creating an SOP for handling Out-of-Specification results.
Purpose
Out of Specifications (OOS) Handling Procedure Scope
Responsibilities
- It is the responsibility of all laboratory personnel working in QC to ensure that this procedure is performed as described and that proper supportive documentation is maintained.
- It is the responsibility of QC Analyst to inform out of specification results to the QC Manager or Supervisor.
- It is the responsibility of QC Manager or Supervisor and Analyst to complete OOS Form and investigate the cause of OOS.
- It is the responsibility of QA to issue OOS tracking number and allow to further investigate for root cause.
- QA ensures that this SOP is followed in its entirety.
- It is the responsibility of QA Head or Designee to approve this SOP.
- It is the responsibility of Director Operation to authorize this SOP.
Definitions & Abbreviations
- Out of specification (OOS) Results: A test that is valid, but the result does not comply with acceptance criteria or the established specifications.
- Retest: Repetition of a test(s) on another aliquot of the same sample or another container (e.g., vial) from the initial sample.
- Sample: The quantity of material taken according to an established sampling plan (e.g. item specifications, stability protocols) that is considered to be representative of the entire batch of material under test.
- Analyst A: A qualified and trained technical person performing the 1st test on 1st sample.
- Analyst B: A qualified and trained technical person with at least equal or more experience than the Analyst A.
- Analyst C: A qualified and well trained technical person with more experience and training than Analyst A and Analyst B. Analyst C should be the subject matter expert.
- Assignable Cause: A documented and scientifically justified determination that an anomalous result can potentially be traced to an error in the laboratory or in processing.
- Un-Assignable Cause: When the cause of test failure cannot scientifically be determined.
- Resample: A new sample taken from the collection of the batch. A resample is warranted when it is demonstrated that the original sample is neither representative nor homogeneous, is contaminated, or has been exhausted/consumed in testing.
- Reference Sample: A sample solution used for comparison purpose during the investigation, which is re-measured and/or reworked parallel to the sample solution in question. Initial results obtained from the control sample should be reliable and as expected from the testing performed.
- Material Review Board (MRB): A team comprising of Head of Quality Assurance, Quality Assurance Manager, Quality Control Manager, Production Manager and Material Manager Head to resolve the issues related to the quality of materials and its impact on the product quality.
- SOP: Standard Operating Procedure
- QM: Quality Management
- OOS: Out of Specifications
- cGMP: Current Good Manufacturing Practices
- HOD(s): Head of Department(s)
- QA: Quality Assurance
- QC: Quality Control
- PR: Production
Procedures
Out of Specifications (OOS) Handling Procedure
- Type 1: Laboratory Error e.g. analyst error, incorrect calculation, malfunctioning of equipment, use of incorrect standards or sample preparation, lack of precision and mis-measurement etc.
- Type 2: Non-process related or operator error in manufacturing i.e. human or mechanical error, which occur during manufacturing e.g. failure to add a component, malfunction of equipment or cross contamination.
- Type 3: Process or manufacturing error due to poor control over processes e.g. incorrect mixing, heterogeneity of blends etc. Confirmation that this was the cause of OOS error will constitute a product failure.
- Type 4: If no process error / Laboratory error is observed during the investigations and results of the tests, constitute a product failure.
OOS Tracking Number
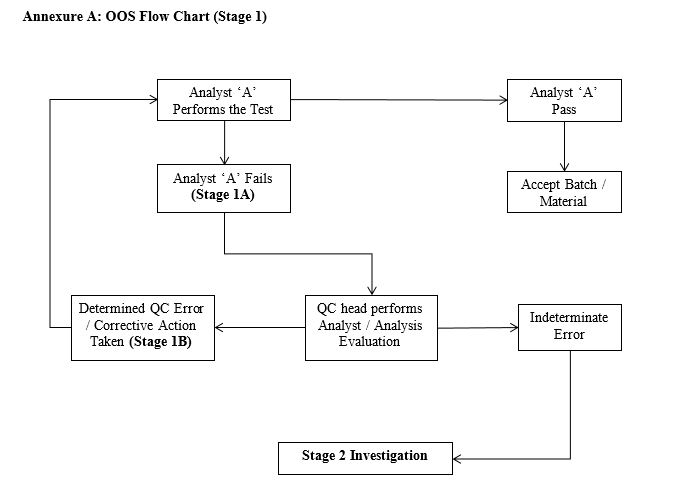
Stage 1: OOS Investigation (Annexure A)
- While carrying the test by Analyst “A”, if the result of test is found out of specification, analyst is to complete “General Section” of form QA/FRM/0000/00, record the result (Stage 1A) and preserve sample, standards and dilution.
- The analyst must inform QC Head / Designee of the OOS result immediately after confirmation of the occurrence.
- Analyst and QC Supervisor jointly complete investigation section of Form QA/FRM/0000/00.
- If any error is found upon investigation (assignable cause), correct the error and same sample should be reanalyzed by the same analyst “A”. Enter result in Stage 1B investigation.
- If the result meets specification, consider test passed and release the material.
- If the result of investigation is indeterminate (un-assignable cause), proceed to Phase II OOS investigation (Stage 2).
- In case the knowledge of Analyst A is not satisfactory, replaces the analyst.
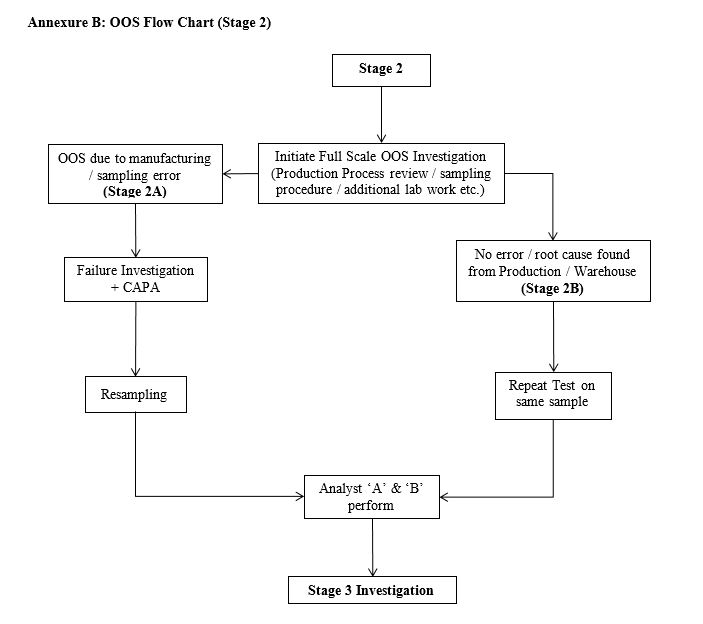
Stage 2: OOS Investigation (Annexure B)
- Full scale investigation Stage 2 is performed from production / warehouse to find out if the factor causing OOS is a result of deviation / manufacturing error / sampling error.
- If the root cause is found to exist from manufacturing / sampling the Stage 1 QC test will be declared invalid and resampling is performed.
- Risk assessment and CAPA are generated to conclude the problem.
- If no error or root cause is found from manufacturing / sampling then retest from the same sample is performed.
- Allow the test to be performed by Analyst “A” and “B” either on the same sample or on resample.
- Forwards the results of Analyst “A” and “B” to Stage 3 investigation.
Stage 3: OOS Investigation (Annexure C)
- Scenario 1: Analyst “A”, FAIL and Analyst “B”, FAIL
- Indicate product / material fails specifications: Reject the material.
- Scenario 2: Analyst “A”, PASS and Analyst “B”, FAIL or results difference is above 3% from each other.
- Allow the test to be performed by Analyst “C”. If result difference is within 3% of pass result, accept material. If fail or result difference is above 3%, indicate product / material fails specifications.
- Scenario 3: If analyst “A” FAIL and Analyst B, PASS or results difference is above 3% of each other)
- Allow the test to be performed by Analyst “C”. If result difference of Analyst C is within 3% of pass result of Analyst B, accept material. If fail, indicate product / material fails specifications.
- Scenario 4: If Analyst “A” PASS and Analyst “B” Pass, Accept the material, if results difference are within 3% of each other.
- Out of Specifications (OOS) Handling Procedure
- In case of Scenario 2 and 3, Analyst “C” performs test on the same sample tested by Analyst A and B.
- If test passes, Analyst “C” repeats test on the same sample used in Stage 1 (results should be within 3% of the previous results).
- QC Head / Manager is to record the result of investigation in Stage 3 of Form (QA/FRM/0007/01) and report results.
- QC Head / Manager gives the assignable root cause at the end of investigation (if any), then it is forwarded to MRB.
MRB Investigation Out of Specifications (OOS) Handling Procedure
- If material fails or there is a failure to give an assignable cause to failure of test at Stage 1, 2 or 3, investigate on the investigation section of the Form (QA/FRM/0000/00).
- State the Formal investigation indicates the investigation is forwarded to the MRB for further evaluation of possible root cause and for determination of corrective action or preventive action.
- If the material fails specifications, E-mail notification is sent to all the concerned departments.
- The MRB will make a decision regarding quality / regulatory impact and disposition of the material(s) / batch(s) and complete the Formal investigation (QA/FRM/0000/00).
- Obtain result on the investigation, conclusion Form (QA/FRM/0000/00).
- OOS is finally closed and archived by QA.
- In case OOS is not closed successfully, MRB will take the final decision for further proceedin
Attached Documents (Forms / Annexure)
- Flow Diagram for Handling OOS (Stage 1) (Annexure A)
- Flow Diagram for Handling OOS (Stage 2) (Annexure B)
- Flow Diagram for Handling OOS (Stage 3) (Annexure C)
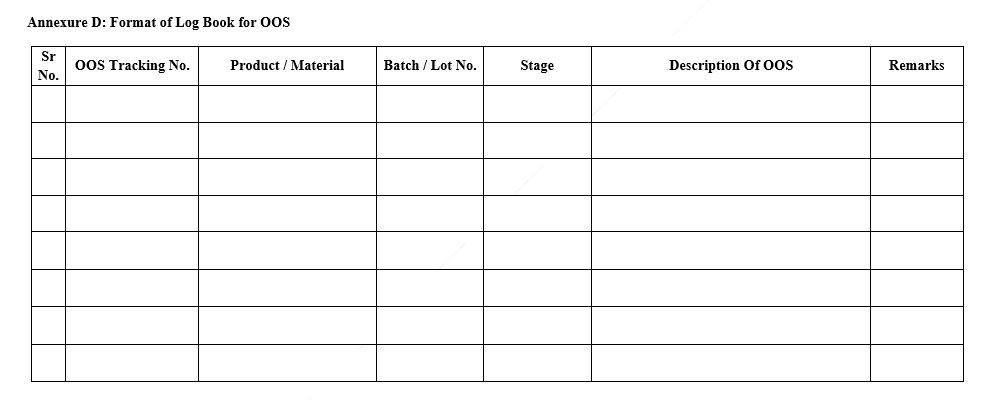
- Format of Log Book for Out of Specifications (Annexure D)
- OOS Investigation Report (QA/FRM/0000/00)
- Out of Specifications (OOS) Handling Procedure
Reference & Linked Documents
- FDA Guidance for Industry: Investigating Out of Specification (OOS) results for Pharmaceutical Production
- WHO: Good practices for national Pharmaceutical control Laboratories
- SOP for Management of Documents
- SOP for Change Control
- SOP for Quality Risk Management
- SOP for Corrective Action & Preventive Action
Revision History
| Revision No. | Date | Responsible Person /Designation | Description of Change |
| 01 | 15.02.22 | QA Manager | To update the SOP as per requirements |
| 02 | 28.05.23 | QA Head | To update the SOP and form as per “SOP for Management of Documents” and Major Flow changes as per Document Change Request |
| 03 | 17.07.23 | QA Head | To update SOP as per discussion with Auditor, flow changes in stage 2 and stage 3, relevant changes in flow diagram and OOS form |
Annexure D: Format of Log Book for OOS
| General Section
Sample Name: ______________________________ SOP number: ________________________ QC Head Sign / Date: ________________________ |
|||
| Stage 1A: Analyst A, First Test Test Result (first test): ___________ Specification: _______________Does not conform QC Investigations: The following step should be taken as part of supervisory assessment. Out of Specifications (OOS) Handling Procedure Details of investigation Yes No·
Remarks of QC Head: Satisfactory Not satisfactory Stage 1B: Analyst A, Second Test (In case of assignable cause) Retest Result (second test):______________
|
|||
Resampling The material is allowed to be re-sampled with the authorization from QA. Sample Quantity: ____________________ Authorized By/Date: _______________ |
|||
| Stage 3: OOS Investigation Date of Re-test: ______________________ Analyst A Name: _____________________Analyst B Name: __________________ Result of Analyst A: _______________ Result of Analyst B: _________________________ Results are within 3% limit: Yes No
Assignable Root Cause (if any): _________________________ Analyst A: ____________________ Analyst B: ____________________ Analyst C: ____________________ QC Head: ____________________ |
|||
MRB Investigation List of Participants:
Investigation by MRB: _______________________
OOS Closed Successfully: Yes No Quality Assurance: ___________________________ |