

A Standard Operating Procedure SOP for Handling of Product Complaints is crucial in the pharmaceutical industry to ensure timely and effective resolution of customer complaints and to meet regulatory requirements.
Purpose
SOP for Handling of Product Complaints Scope
Responsibilities
- It is the responsibility of distribution manager to forward the complaint received from distributor, retailer or customer to QA .
- In case of internal complaint, Head of Department is responsible to forward the complaint to QA.
- It is responsibility of QA to coordinate and process all product complaints including product complaints related to Adverse Drug Reactions
- It is the responsibility of QA to send final reply of the complaint to complainant and final disposition of all product complaints.
- Regulatory Affairs department is responsible for coordinating, processing and responding to all product complaints related to or from regulatory agencies.
- QA ensures that this SOP is followed in its entirety.
- SOP for Handling of Product Complaints
- It is the responsibility of Assistant QA Manager or Designee to review this SOP.
- It is the responsibility of QA Head or Designee to approve this SOP.
- It is the responsibility of Director Technical / Operations to authorize this SOP.
Definitions & Abbreviations
- Product Complaint: Any complaints related to quality, efficacy, purity, identity and stability of products which may or may not affect the health of the customer
- Classification of Complaints: There are three classification of complaints, critical, major & minor.
- Critical: Process performed incorrectly; product quality, purity or safety affected, e.g. label mix-ups, wrong active pharmaceutical ingredient, wrong strength, overdosing or some critical side effects or adverse drug reactions which may cause the death of the patient or permanent disability.
- Major: Process parameters are incorrect, product can cause illness or mistreatment; product quality, purity or safety in question which may put patient at some risk but not life threatening e.g. out of specifications, broken / defected tablets, microbial growth, contamination, impurities, leakage, missed overprinting, wrong price printing etc.
- Minor: Brief excursion from specifications which are not life threatening and not directly affecting product quality, purity or safety e.g. shortage or excess quantity, empty pockets, improper sealing, dirty unit packs, missing leaflets etc.
- External Complaints: Complaints reported by the Distributors, Retailers, Consumers, Doctors, Hospitals and Ministry of Health about those finished products that have been marketed.
- Clinical Complaints: Complaint reported by physician, pharmacist or patient related to safety of Pacific Pharmaceuticals product.
- Non – Clinical Complaints: Complaints reported by physician, pharmacist or patient, related to Pacific product design, including product insert package, color, text, and size, suitability, condition or the appearance of the package, label or the dosage form that might considered to reduce the usefulness of the product.
- Regulatory Complaints: Complaint reported by Ministry of Health related to regulatory requirement of Pacific product or the design that might be considered to reduce the usefulness of the product.
- SOP: Standard Operating Procedure
- QM: Quality Management
- CAPA: Corrective Action & Preventive Action
- FG: Finished Goods
Departmental Codes
- QA: Quality Assurance
- QC: Quality Control
- PR: Production
- WH: Warehouse
Procedures
- Complaints are received from internal or external source and as verbal feedback or written feedback.
- Verbal feedback is received in person or via a telephone conversation.
- Written complaints are received in the form of letters, faxes, e-mails, Nevertheless, all complaints are recorded using the Product Complaint Form (QA/FRM/000/00)
Handling & Investigation and Reporting of Internal / External Complaint
- All external complaints are received by QA either via Distribution Manager or directly to the plant.
- Complaint either external or internal is forwarded to QA within 24 hours from when the complaint was received on “Product Complaint Form” (QA/FRM/0012/01) along with the complaint sample for evaluation and possible investigation.
The case should contain the following information.
- Name and Address of the complainant
- Complainant Area
- Date of Complaint
- Product Name
- Batch No.
- Mfg. Date
- Exp. Date
- Quantity
- Pack size
- Description of Complaint
- SOP for Handling of Product Complaints
- QA assesses the validity of the complaint and need of further investigation and evaluation.
- QA calls the meeting of Technical Committee for complaint root cause review and investigation.
- Technical committee comprises of Director Technical / Operations, QA Head, QC Head, Production Head, Regulatory Head (if required), Procurement Head (if required), Warehouse Incharge are included in technical committee.
- Technical committee reviews and determines the likely root cause of complaints as product complaint is quality related, clinical related, safety related, storage problem, transportation problem or other categories by reviewing the batch processing record and other records of documentation of the said product.
- If required, QA requests QC to perform required test on retained samples of the effected batch of product and other batches in order to determine whether they are also affected (In particular, other batches which may contain reworks of the batch under investigation).
- If the QC test indicates quality or safety related problem, the case is sent to the Recall.
- Committee to proceed as per SOP for Recall as soon as possible.
- If an investigation reveals no quality related issues, QA takes corrective / preventive actions to rectify the problem.
- QA determines the effectiveness of the CAPA taken and completes the Product Complaint Form.
- QA prepares a case investigation report regardless of the nature of complaints outlining the course of investigation including any test result and the corrective action and sends to Distribution Manager to be communicated to the complainant.
- A copy of report is archived with the product complaint file at the QA.
- QA completes its investigation within 15 days from when the complaint was received.
Complaint Records
- Complaint record includes ‘‘Drug Product Complaint File’’ containing.
- Product Complaint Form (QA/FRM/0000/00).
- Respond to complainant with Copy of investigation report.
- Complaint records are kept for one year after the expiry of the product.
- On the basis of complaint if CAPA is required enter complaint No. on CAPA form QA/FRM/0000/00 as well as on change control form QA/FRM/0000/00.
Attached Documents (Forms / Annexure) SOP for Handling of Product Complaints
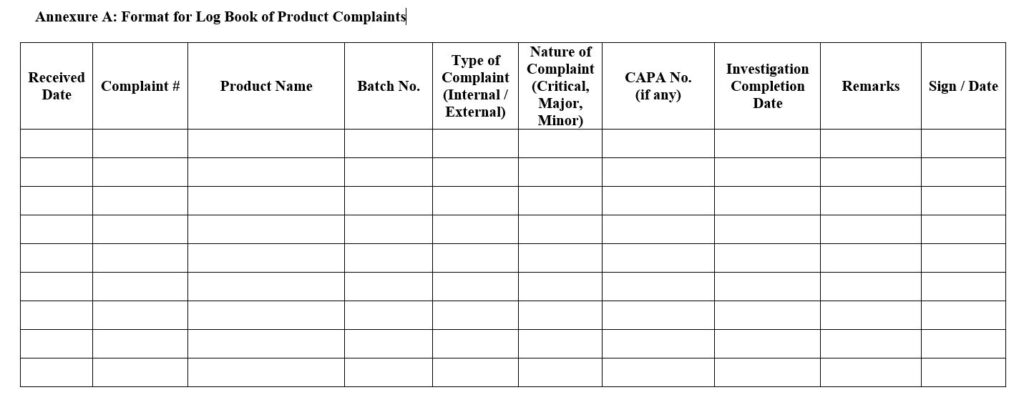
- Format for Log Book of Product Complaints (Annexure A)
- Product Complaint Form (QA/FRM/0000/00)
Reference & Linked Documents
| SECTION A: To be Filled by the Individual Reporting the Complaint
Date of Complaint: ____________ |
|||||||||||
SECTION B: To be Completed by the QA
Comments: _______________________________________________________________________________ |
|||||||||||
|
|||||||||||