
A Standard Operating Procedure SOP for Supplier Qualification of Excipients is essential to ensure the quality, safety, and regulatory compliance of the materials used in pharmaceutical and other industries. Excipients are critical components of drug formulations, and it is crucial to establish a systematic process for qualifying and approving suppliers.
Purpose
- To describe procedure for selection, qualification and approval of Excipient’s supplier including domestic and foreign manufacturer
- To establish a standardized process for the qualification of suppliers providing excipients.
- To ensure the quality, safety, and regulatory compliance of excipients used Pharmaceuticals products.
Scope
- The scope of this document pertains to suppliers of all excipients used in Pharmaceuticals Ltd
- This SOP applies to all excipient suppliers considered for use by Pharmaceuticals products.
- The procedure covers the evaluation, approval, and ongoing monitoring of excipient suppliers.
Responsibilities
- It is the responsibility of procurement department to follow this SOP as it is written
- It is the responsibility of QA to complete Risk Assessment Form
- It is the responsibility of QA Documentation Officer to prepare and update this SOP
- It is the responsibility of QA Manager to make certain that this SOP is followed in its entirety
- It is the responsibility of Assistant QA Manager or Designee to review this SOP
- It is the responsibility of QA Head or Designee to approve this SOP
- SOP for Supplier Qualification of Excipients
- It is the responsibility of Director Technical / Operations to authorize this SOP
Definitions & Abbreviations
- Manufacturer/ Supplier/ Vendor: The all three terms refer to manufacture of excipient
- Excipient: Any added materials, other than API, which will not affect the bioavailability or / and stability of finished product
- SOP: Standard Operating Procedure
- QM: Quality Management
- QA: Quality Assurance
- QC: Quality Control
- GMP: Good Manufacturing Practices
- BSE: Bovine Spongiform Encephalopathy
- TSE: Transmissible Spongiform Encephalopathy
- DMF: Drug Master File
- COA: Certificate of Analysis
Procedures SOP for Supplier Qualification of Excipients
It is a GMP requirement that excipients and excipient suppliers should be controlled appropriately based on the results of a formalised quality risk assessment in accordance with the European Commission ‘Guidelines on the Formalised Risk Assessment for Ascertaining the Appropriate Good Manufacturing Practice for Excipients of Medicinal Products for Human Use’
The level of supervision should be proportionate to the risks posed by the individual materials, taking account of their source, manufacturing process, supply chain complexity and the final use to which the material is put in the medicinal product
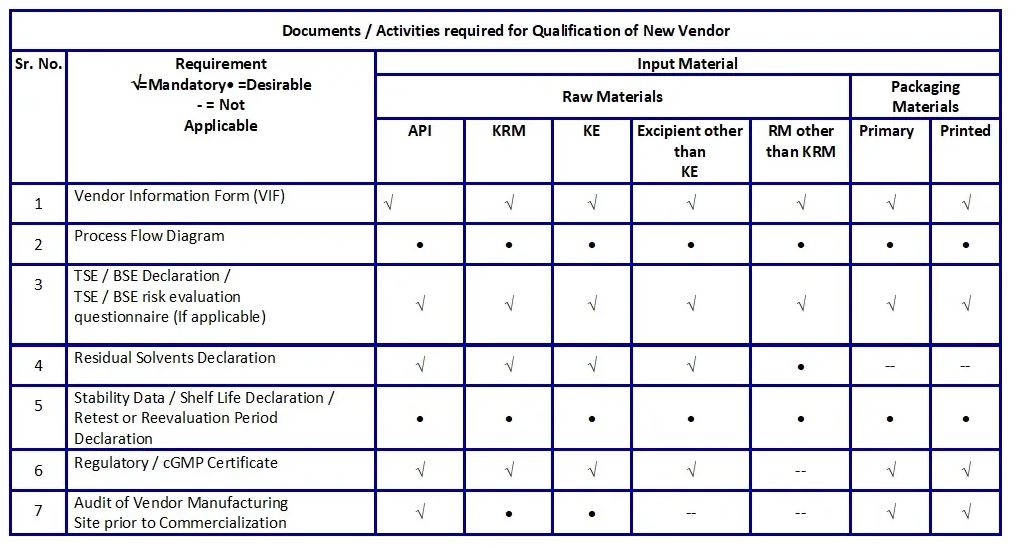
To start with, procurement send out request to manufacturer / supplier receive the following document:
• GMP certificate
• Samples from three consecutive batches
• COA of excipient
If COA is not received or received incomplete, then an alternative supplier must be searched for
If review of COA is satisfactory, samples are transferred to QC for testing
If QC testing is passes, a request is send out to supplier for submission of the following document:
• Site Master File (SMF), if available
• Stability Report of Excipient
• TSE / BSE compliance certificate
The above document is reviewed by QA. The procurement send out “GMP Questionnaire” to vendor for additional information
Risk Evaluation Parameters
- For each excipient, SMF (if available), stability report, TSE / BSE and GMP questionnaire is thoroughly reviewed by QA against the following risk factors listed in “Risk Assessment for Excipients”.
- QA Assessment is in the form of quantitative scoring system from 1-3; numerical value of 1 is indicates LOW RISK, The numerical value of 2 indicates MEDIUM RISK, numerical value of 3 indicates HIGH RISK
- The result of risk evaluation is to be recorded in the “Risk Assessment Summary Result”.
The Risk Categories
- Stability programme
- Route of manufacture – the source (animal, mineral, vegetable) and process (e.g. synthetic, fermentation) must be considered. Areas for consideration include TSE, potential for microbiological contamination, impurities from raw materials (e.g. aflatoxins, pesticides) or generated as part of the process and carried over (e.g. residual solvents and catalysts)
- Potential toxicity / flammable material
- Presence of potential impurities
- Supply change complexity
- Presence of Quality Management System
- Evidence of cross contamination
- Presence of TSE / BSE compliance
- Microbiological testing of material
- Environmental control and storage condition control of temperature, humidity, and differential pressure
- Change management and deviation handling and change control policy
- Transport system, cold chain if any
- Packaging integrity evidence assess on the base of sample received
- Grade of material (e.g. Ph. Eur., Food grade)
- Quality defects of material
- Route of administration of material
- Use of material during manufacturing
- Material affecting quality attributes of product
- Sufficient number of employees in the company
- Defined and written job description of all employees
- Existence of in house training programmes for employees
- Existence of training programmes for daily hygiene practice in the company
- Availability of written procedures for preventive maintenance of all equipment
- Existence of unique coding system for all the raw as well as packaging materials
- Existence of written procedure for vendor qualification
- Existence of independent QC
- Retention of all records for incoming material and excipients along with retained samples of all material for specified time
- Existence of written procedure for complaints and recall
- Existence of written procedure for Deviation and CAPA
- Existence of written procedure for self-inspection
- SOP for Supplier Qualification of Excipients
Risk Ranking for Excipient
Risk score and description of risk for each must be entered in the Form.
Upon completion of risk evaluation, manufacturer must be assigned risk ranking of Low, Medium or High:
Risk score:
• If total score is less than or equal to 40, the risk of using excipient is LOW
• If total score is between 40 – 65, the risk of using excipient is MEDIUM
• If total score in between 65 – 99, the risk of using excipient is HIGH
If risk evaluation is LOW – No site audit is required and the supplier is to be placed on the “Approved Vendor List”.
If risk evaluation is MEDIUM – audit of the vendor is conducted by quality team of Pharmaceuticals as per “Audit Checklist for Qualification of API / Excipients Manufacturer”.
If risk evaluation is HIGH – then supplier is put on “Disapproved Vendor List”.
Continue Monitoring
- Once material is received, it should be critically monitored. Any adverse observation during the period of stability study is immediately brought to the notice of Production, Procurement and QA departments
- Approved vendor are placed on the “Approved Vendor List” Form.
- Disapproved supplier is placed on the Form.
- SOP for Supplier Qualification of Excipients
On Going Review SOP for Supplier Qualification of Excipients
- Once a supplier is approved, the quality of material supplied by them must be monitored on a continuous basis in order to highlight any change in quality
- If there are any issues related to re-testing of excipient, the Vendor Complaint Form is filled by QC and submitted to QA for verification and further processing
- If any shipment / supply rejected by QC continuously for 2 times on quality related issues, vendor / supplier should be black listed from the vendor list and name should be placed in “Vendor Black List.
- Vendor list should be up to date using document change control due to black list of any vendor / supplier
- Once QA has verified problem after the retest performed on the material, the complaint form is forwarded to procurement for delivery to manufacturer
- Copy of Vendor Complaint Form, (QA/FRM/0015/04) is to be archived in vendor file by procurement department
- If complaint not addressed properly by the vendor, material is to be rejected and send for incineration. Further supply is stopped immediately
Note 1: Vendor requalification is to be performed in every three years or sooner as needed
Note 2: Upon risk evaluation vendor is placed either in Approved Vendor List for Excipients or in Disapproved Vendor List.
Note 3: If the vendor resolves issue related to quality then upon confirmation and risk evaluation, it will be placed in Approved Vendor List but in case the problem is faced again, it will be placed in Vendor Black List.
Attached Documents (Forms / Annexure)
- Risk Assessment for Excipients
- GMP Questionnaire
- Approved Vendors List of Excipient Supplier
- Disapproved Vendor List of Excipient Supplier
- Risk Assessment Summary Result
Reference & Linked Documents
- ICH Harmonised Tripartite Guideline Good Manufacturing Practice Guide For Active Pharmaceutical Ingredients Q7, Version 4
- European Commission ‘Guidelines on the Formalized Risk Assessment for Ascertaining the Appropriate Good Manufacturing Practice for Excipients of Medicinal Products for Human Use’
- SOP for Management of Documents
- SOP for Supplier Qualification of API
- Log book of Vendor Qualification
- Quality Audit Check List for Qualification of API / Excipients Manufacturer
- Vendor Complaint Form
- Record of QC Testing Material Vendor Qualification
- Vendor Qualification Schedule
- Vendor Black List Form
- SOP for Supplier Qualification of Excipients
- Vendor Appraisal Audit Report