

In the pharmaceutical industry, Handling of Deviation in Pharmaceutical Industry is a crucial aspect of ensuring product quality, patient safety, and regulatory compliance. A deviation is any departure from approved procedures or specifications, and it can occur at various stages of the pharmaceutical manufacturing process. Managing and investigating deviations effectively is essential to prevent recurrence and maintain a high level of product quality. Here are key steps and considerations in handling deviations in the pharmaceutical industry.
PURPOSE
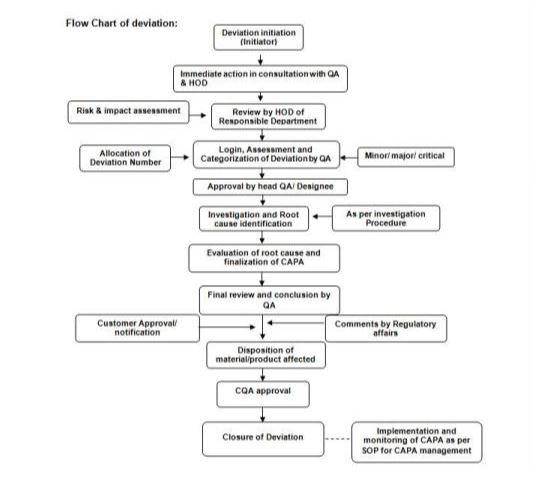
To define a procedure for handling, recording, investigating and evaluating of planned and unplanned deviations from standard procedure, manufacturing instructions and quality related procedures that impact on product quality.
SCOPE of Handling of Deviation in Pharmaceutical Industry
The Procedure applies to the following:
- Product standard and registered details.
- Manufacturing processes/operations.
- Yield and reconciliation limits.
- Facilities including equipment, utilities and critical instrument calibration failures.
- Handling of vendor (supplier) complaints.
- Operation outside validated ranges, registered details or legal requirements.
- Documentation errors that potentially affect product quality or could lead to incorrect release of a batch.
- Diverse trends identified from data analysis.
- Handling of Deviation in Pharmaceuticals Industry
- Environmental action and alert limit e.g. Temperature, Humidity, Particle counting, differential pressure etc.
- To ensure that all events and deviations are recorded & assessed.
- To ensure appropriate immediate actions are taken.
- To identify the root cause through investigation.
- To ensure action arising from the review is recorded in a formal manner.
- Corrective and preventive actions (CAPAs) are applied to prevent recurrence in originating area, and occurrences in other areas where there is a potential for the problem to occur.
ROLES & RESPONSIBILITIES
| Roles | Responsibilities |
| The Observer | To record the deviation and notify the manager of deviation. |
| Departmental Manager/ Investigation Leader |
|
| Quality Assurance Manager |
|
PROCEDURE
Department manager and Quality Assurance Manager will in conjunction decide whether deviation planned or unplanned.
Unplanned Deviation:
- While carrying out day-to-day activities, there are chances of unplanned deviations (unforeseen deviations) to occur.
- Person who discovers the deviation must record the detail of deviation /event in the section.
- The departmental manager will sign in the deviation record form and take the immediate action e.g. stopping operations, segregating material.
Initial Assessment of Deviation:
- The departmental manager gathers the relevant data for assessing the impact of the deviation. (If during impact assessment any of impact is identified take action accordingly)
- The departmental manager (s) should record the initial assessment.
Investigation and Identification of Root Cause & CAPA Objective:
- It is the responsibility of the departmental manager where the deviation has occurred to nominate an investigation leader.
- The investigation leader will nominate the team member on the basis of following:
- Technical knowledge of the process.
- Knowledge of GMP.
- Knowledge of relevant regulatory requirements.
Approve Conclusion of Investigation and Raise CAPA:
Deviation is considered completed if:
- Investigation completed and root cause identified.
- Classification of deviation has been documented on the basis of finding of the investigation.
- CAPA has been identified & approved.
- Handling of Deviation in Pharmaceutical Industry
- Based on the initial assessment, investigation, identification of root cause, implementation of actions & identification & approval of CAPA action plans, QA Manager may take the decision to release material / product implicated by the deviation.
Planned Deviation:
- Any Deviation in manufacturing process / procedure / testing procedure / operating / calibrating procedure, batch size, equipment, operating environment or any other GMP critical process which is pre-planned for the forthcoming batches, shall be known as planned deviation.
- The concerned department executive / manager shall initiate the “Planned Deviation Process” by documenting the details in the deviation record form.
- Concerned Manager and QA Manager shall review the proposal for planned deviation, justification given and compliance to regulatory requirements, for necessity / feasibility of the same and sign off in the specified column.
- QA shall assign a unique number to each planned deviation.
- QA Manager shall review the planned deviation with respect to impact on product quality, necessity / feasibility of the deviation proposed, rationale / justification & compliance to cGMP / regulatory requirements, along with the adequacy of the supporting data attached and approve or reject the deviation.
- If approved, the deviation shall last for a defined number of batches or number of days.
- Date of closing or stoppage of such Planned Deviations must be recorded too.
Control Handling of Deviation in Pharmaceutical Industry
- Review of Performance:
It is the responsibility of the departmental manager reviews of all deviations, their related investigations and effectiveness of the resultant CAPAs. - Monthly Reporting:
QA Manager will report all the deviations on the monthly basis. - Numbering of Deviation:
QA department will allocate the reference number.
Example DEV/YYYY/00
| DEV | YYYY | 00 |
| Deviation | Year | Sequential no. |
Document Filing:
- Deviation copy must be a part of the batch record and APR. The Original record must be maintained in the office of the QA Manager.
- Timeline within 30 days. Report for toll manufacturing.
- Timeline for investigation should be closed and singed of within 20 days.
Interim Report:
- Interim records are required for critical and non-critical deviations if the due date for QA decision cannot be adhered to.
- In case of extension, concerned client must be informed.
- Interim report must be approved by QA and CEO.
Interim reports must include the following information:
- Current status of the deviation, including action taken in the past 30 calendar days.
- Justification / rationale for extending the investigation an additional 30 calendar days.
- Ongoing and planned actions, including timelines and responsibilities for the actions.
- Responsibility for follow up-monitoring and ongoing reporting and notification.
- Estimated timeline to finalization of investigation and closure of deviation.
Retention deviations:
Deviations are retained as per the defined procedure. Handling of Deviation in Pharmaceutical Industry
Soft copy will be retained for lifetime and hard copy will be retain for 3 years.
CLASSIFICATION OF DEVIATION:
| Classification | Description | Expected Consequences |
| Critical Deviation | A critical GMP deviation could endanger product / patient safety. Failure of detection would lead to recall. Critical deviation will be observed during the process check or controls. | Critical deviation Will results in.
|
| Major Deviation | A major GMP deviation/exception does not directly influence product safety and efficacy but may require immediate action before approval is granted to manufacture or continue to manufacture. | This could lead to:
|
| Minor Deviation | A minor GMP deviation/exception does not directly influence product safety and efficacy but may have an impact on cosmetic quality. This also includes minor document irregularities. | This could lead to:
|
REFERENCE
ICH Guideline Q7, point # 2 – Quality Management.